Ongoing Research projects
Spiral ganglion neurons (SGNs) are bipolar primary neurons in the cochlea that transmit auditory information from the sensory hair cells in the organ of Corti to the brain. Loss or dysfunction of SGNs degrade auditory processing leading to sensorineural hearing loss that affects millions of people. Such loss of SGNs, and their peripheral axons can occur after hair cell death, or after the loss of their synaptic connections (termed as ribbon synapse) with hair cells caused by exposure to noise, ototoxic drugs, or aging. Functional SGNs are critical for positive outcomes with hearing aids or cochlear implants and are necessary to fulfill the promise of future hair cell regeneration strategies. Thus, there is a need to develop methodologies that could be used to maintain, repair, and regenerate SGNs. However, the mechanisms of SGN degeneration and survival are poorly understood. Our long-term goal is to determine the biological mechanisms of SGN degeneration, repair and survival to develop therapies that maximize hearing aid and cochlear implant technologies to treat hearing loss.
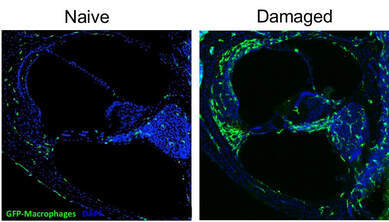
We are investigating innate immunity to hearing loss and SGN degeneration, survival, and repair. Cochlear damage is associated with an increase in macrophage (innate-immune cells) numbers (see figure on right) but, their exact contribution to cochlear toxicity and hearing loss are unclear. We have identified fractalkine signaling as a key positive regulator for SGN survival and synapse repair. Fractalkine signaling is a unique immune-neuron receptor-ligand pair where the ligand, fractalkine (CX3CL1), is a transmembrane cytokine which is constitutively expressed on SGNs and binds to its exclusive receptor, CX3CR1, expressed by innate-immune cells such as macrophages and monocytes. We have demonstrated that hair cell death or synaptic damage can recruit macrophages into the inner hair cell-synaptic region and spiral ganglion. Lack of CX3CR1 exhibits impaired spontaneous synaptic repair and greater SGN loss than CX3CR1-positive controls. This suggest that macrophages can play a neuroprotective role to promote SGN survival and synaptic repair through fractalkine signaling. A more complete understanding of how macrophages and fractalkine regulate neuron survival and synaptic repair will provide critical information towards developing novel immunotherapies to prevent auditory nerve degeneration or promote nerve regeneration and re-establishment of their synaptic connections and restore hearing.
We are investigating innate immunity to hearing loss and SGN degeneration, survival, and repair. Cochlear damage is associated with an increase in macrophage (innate-immune cells) numbers (see figure on right) but, their exact contribution to cochlear toxicity and hearing loss are unclear. We have identified fractalkine signaling as a key positive regulator for SGN survival and synapse repair. Fractalkine signaling is a unique immune-neuron receptor-ligand pair where the ligand, fractalkine (CX3CL1), is a transmembrane cytokine which is constitutively expressed on SGNs and binds to its exclusive receptor, CX3CR1, expressed by innate-immune cells such as macrophages and monocytes. We have demonstrated that hair cell death or synaptic damage can recruit macrophages into the inner hair cell-synaptic region and spiral ganglion. Lack of CX3CR1 exhibits impaired spontaneous synaptic repair and greater SGN loss than CX3CR1-positive controls. This suggest that macrophages can play a neuroprotective role to promote SGN survival and synaptic repair through fractalkine signaling. A more complete understanding of how macrophages and fractalkine regulate neuron survival and synaptic repair will provide critical information towards developing novel immunotherapies to prevent auditory nerve degeneration or promote nerve regeneration and re-establishment of their synaptic connections and restore hearing.
Project 1: Contribution of resident and infiltrated macrophages towards neuron survival in damaged cochlea
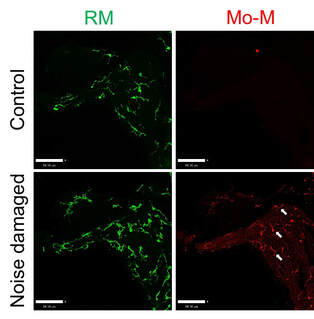
Damage to the cochlea is associated with activated resident macrophages (RM) and infiltration of monocyte-derived macrophages (Mo-M) from the vasculature. It is unclear whether both of these macrophage populations are differentially neuroprotective. We are using RM and Mo-M specific mouse reporter lines to characterize the spatiotemporal distribution, morphological and molecular phenotype of macrophages following damage. We are also independently depleting the two macrophage populations to determine their effect on SGN degeneration and survival. These studies will reveal the heterogeneity in cochlear macrophages in damaged cochlea and shed light on the specific roles of CX3CR1-expressing RM and Mo-M in SGN survival.
Project 2: Role of CX3CR1 as regulator of macrophage response in damaged cochlea
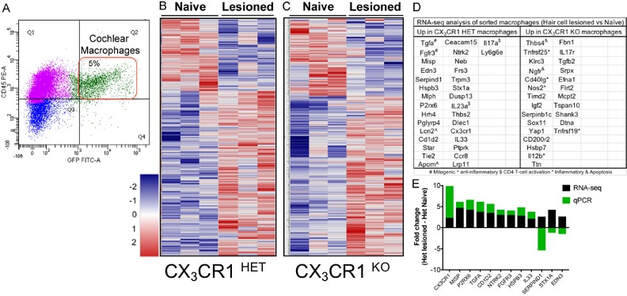
We have reported that lack of CX3CR1 is associated with increased SGN loss in damaged cochlea. Yet, the precise molecular mechanisms for such neuronal loss are unknown. Using transcriptomic and proteomic research tools we are profiling SGNs and cochlear macrophages with intact fractalkine signaling and that lack CX3CR1 with and without damage. The data from these studies will allow us to identify critical pathways and candidate genes that are differentially expressed in CX3CR1-deficient macrophages that regulate SGN degeneration and survival following cochlear damage.
Project 3. Association of human CX3CR1 polymorphisms in sensorineural hearing loss
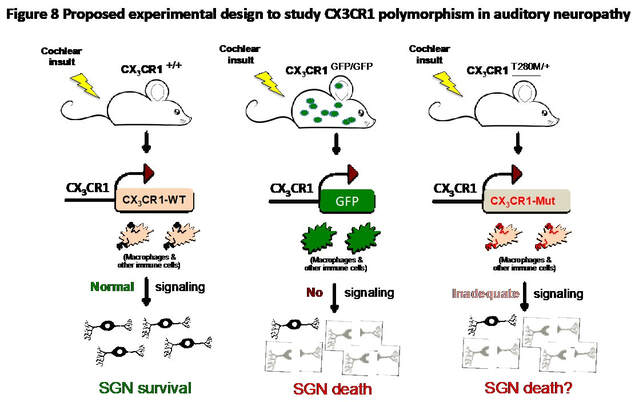
Approximately 25-30% humans carry single nucleotide polymorphisms (SNPs) in the CX3CR1: Val249Iso and Thr280Met. These variants lead to defective receptor-ligand binding and are associated with multiple neurodegenerative diseases. Based on known heterogeneity in human SGN loss and our observation of increased SGN loss in CX3CR1-deficient damaged cochlea, it is of significant clinical relevance to understand the effects of the normal human CX3CR1V249/T280 receptor and its polymorphic variant CX3CR1I249/M280 in cochlear pathology and hearing loss. Using a novel mouse line expressing these human CX3CR1 SNPs we are investigating the effect of I249/M280 polymorphisms in cochlear function, and structure following damage. These studies may identify a novel immune-related gene SNPs that can increase the risk of hearing loss.
Project 4. Role of Macrophages towards Damage and Repair of Cochlear Ribbon Synapses
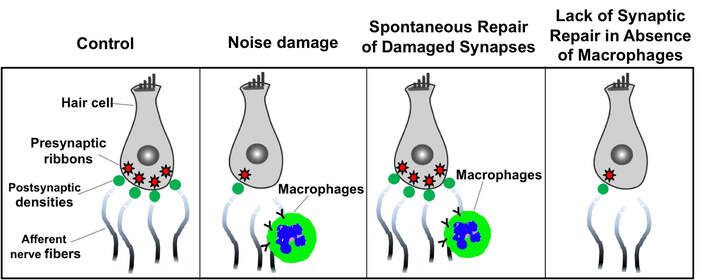
We recently demonstrated that cochlear resident macrophages migrate into the damaged IHC-synaptic region immediately after synaptopathic noise trauma and directly contact the damaged synaptic connections. However, the functional consequences of these contacts and mechanisms of macrophage migration towards damaged synaptic region are unknown. To address this, we are eliminating cochlear macrophages to examine their impact on damage and spontaneous repair of ribbon synapses following noise trauma. We are also examining whether macrophage repopulation in the cochlea lacking macrophages repair noise-damaged ribbon synapses. In addition, we are determining the molecular mechanisms of macrophage migration into the noise-damaged synaptic region and spiral ganglion and how macrophages regulate repair of damaged ribbon synapses. Together, the outcomes will advance our understanding on the role of macrophages in cochlear pathology and identify a novel non-sensory cellular mechanism to preserve or repair cochlear synapses that can be harnessed to develop immunotherapies to treat hidden-hearing loss or cochlear synaptopathy-related tinnitus or hyperacusis in affected individuals.
Project 5. Fractalkine Overexpression in Damage and Repair of Cochlear Ribbon Synapses
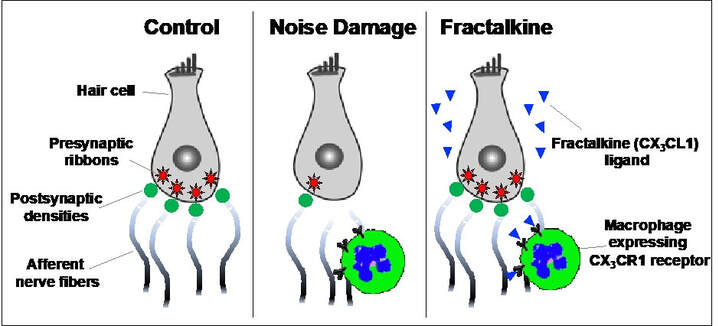
Our data imply that intact fractalkine signaling is necessary for synaptic repair and neuron survival in the damaged cochlea. However, it remains unclear whether fractalkine signaling is sufficient to prevent loss of, or repair damaged, synapses due to noise trauma or excitotoxicity. Moreover, the precise contribution of soluble and membrane isoforms of fractalkine ligand (CX3CL1) in preventing synaptic degeneration or initiating repair remains to be determined. The goal of this project is to test the efficacy of overexpression of CX3CL1 ligand in preventing loss of, or repairing damaged, IHC ribbon synapses. The data from these studies will potentially implicate a new target, fractalkine, to preserve or restore synapses and increase SGN survival in the damaged cochlea. Such results will enable the development of novel fractalkine-based immunotherapies to benefit individuals with auditory synaptopathy and SGN loss due to either noise trauma, ototoxic medications, aging or tinnitus when associated with loss of synapses.
